 V and C region bearing Antibody Web Interface
V and C region bearing Antibody Web Interface
Last updated: 19/08/2024
Example of unpaired sequence
Example of paired sequence
AIRR example
Cell ranger example
customized repertoire file
Download all the entris in VCAb here:
Download all entries in VCAbDownload the all the numbered sequences in VCAb here:
Download all the numbered sequences in VCAb ![]()
V and C region bearing antibody Web Interface
VCAb is a web application to investigate antibody isotypes structural features, bridging antibody sequence repertoire acquired via next generation sequencing to the available antibody structural space of structures covering both V and C regions. Antibody structures in VCAb are clearly annotated with isotype, light chain type, structural coverage, information (such as interface residues, CH1-CL torsion angles) related with CH1-CL interface, and in silico mutational scanning profiles.
VCAb is now available online. Users can search for antibodies of their interest, then view and inspect annotations and structures of them interactively.
If you are interested in the generation process of available experimental structural space of antibodies with both V and C regions, please go to Antibody structural space.
The layout of VCAb webpage
As indicated in the navigation bar, four pages are available in VCAb.

The search page
In this page, the user can query the database, view the antibody information and inspect the antibody structures interactively. The search page can be divided into 3 panels: the Query Input panel, Antibody Information panel, and Interactive 3D Structure Viewer and Sequence Viewer (Please click on the corresponding link for details).

The statistics page
This page summarizes the statistics of available antibodies in current released version of VCAb. You can view the statistics by different standard: species, isotype, light chain type and structural coverage. Multiple standards can be selected at the same time.

The download page
In this page, you can download the annotations for all the antibodies in VCAb. All the sequence in VCAb of both V and C regions are numbered under IMGT scheme, using ANARCI_vc, a package modified from ANARCI to enable both V and C numbering. All the numbered sequence in VCAb can be downloaded here.
The documentation page
This page is a copy of this github wiki.
The about page
This page included a brief introduction of VCAb web tool. Way to cite VCAb is also provided here.
- Query by PDB ID
- Query by antibody features
- Query by sequence
- Query by CH1-CL interface similarity
- Query by uploading repertoire file
Query by PDB ID

Query by antibody features

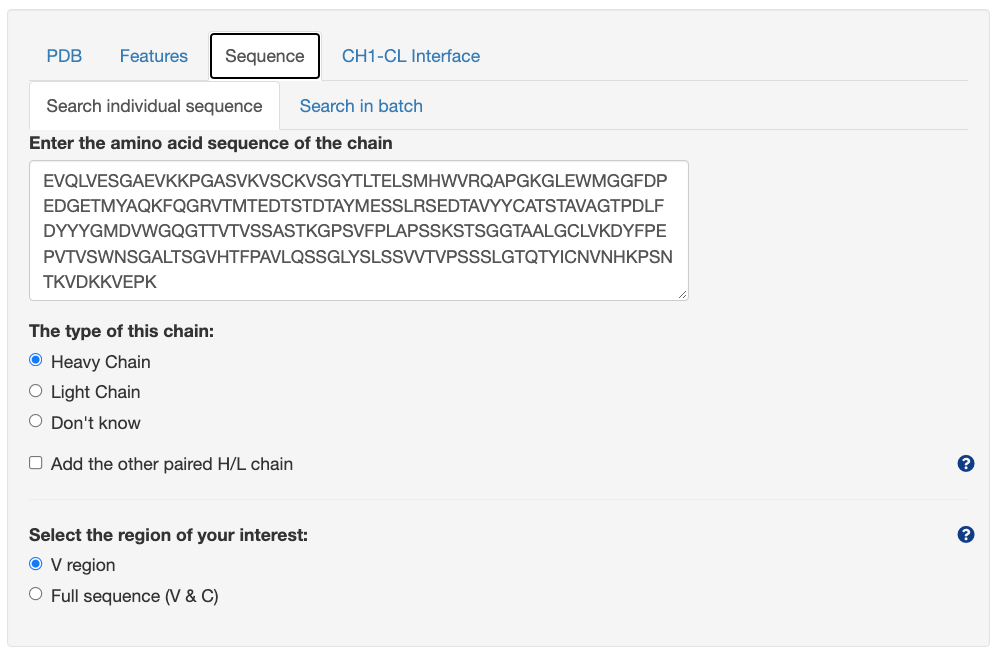
Query by sequence
Search individual sequence
Unpaired sequence
The structural hits will be ranked by the Percentage Identity to the inputted sequence.
Paired sequence

The structural hits will be ranked by considering the average percentage identity of both heavy and light chains inputted. Percentage identity to both heavy and light chain will be listed separately.
Search sequences in batch

User can upload the fasta file containing multiple sequence for search (200 seqs maximum). Example fasta files for both paired and unpaired searching mode are provided to the user, user can click the link to load the example file and search.
Note: In the paired mode, in order to indicate the paired heavy-light chain sequences, the title for the fasta sequence must be in this format: AbName-HorL, where AbName is the name for the H-L pair, HorL can only be the letter of "H" or "L" (case sensitive). The paired sequence must contain the same AbName, with only one H chain and one L chain.
Query by CH1-CL interface similarity


Query by uploading repertoire file

Upload Files
User can upload the repertoire files to VCAb through the following methods: (Please note: to re-upload the repertoire file, please click the "Clear uploaded repertoire file/url" button to clear the previous uploaded file first.)
- Click the "Browse..." button to upload file from your local computer
- Paste a http address of the repertoire file to the box of "Or input a http address for repertoire file here:"
- Send the result calculated by BRepertoire in the "property calculations" panel of the "Analysis" page to VCAb.
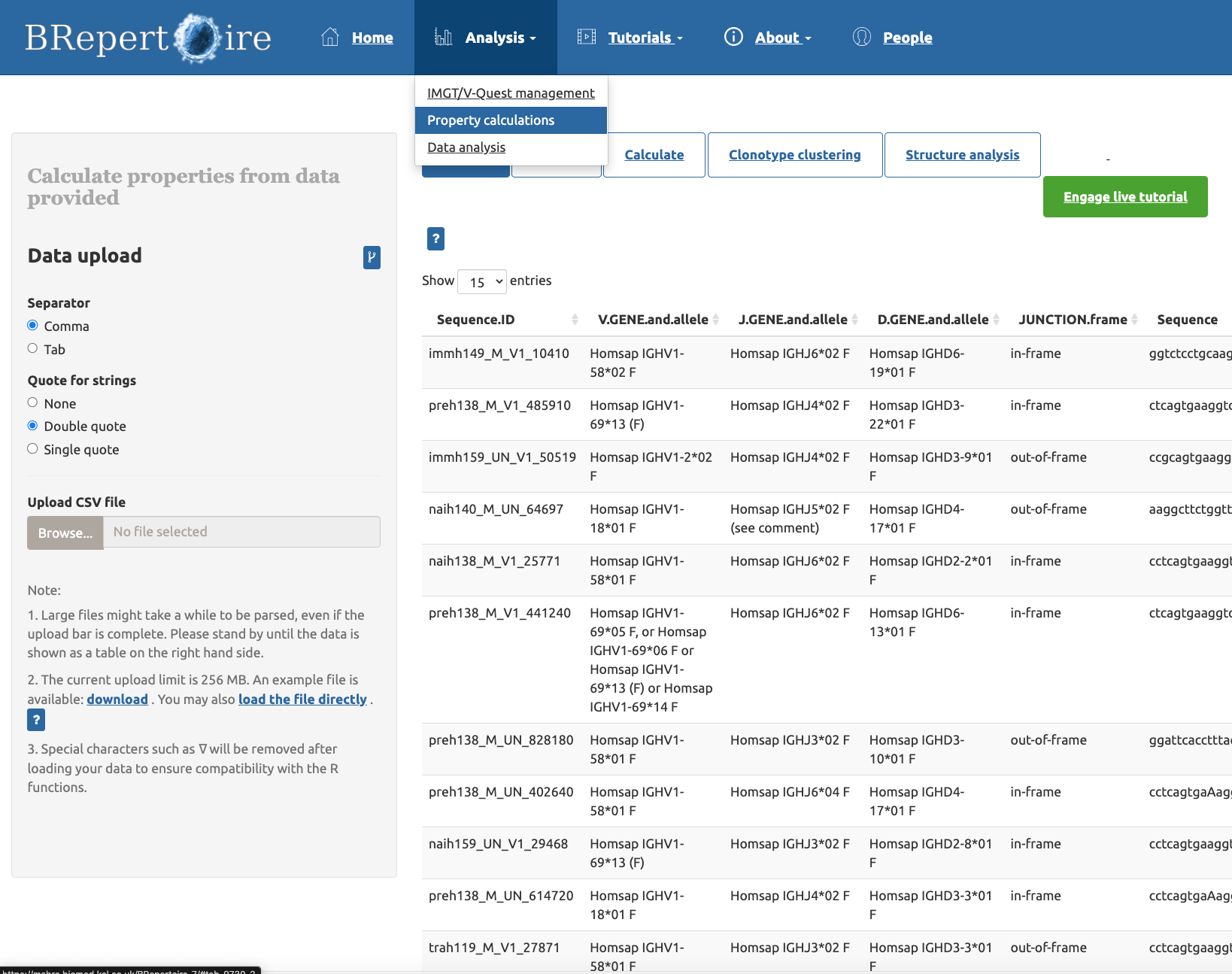
Figure above: The "property calculation" panel of the "Analysis" page

Figure above: Click the "Structure analysis" option, and click the button of "Send to VCAb for mapping to antibody structures" to send the table from BRepertoire to VCAb.

Figure above: The BRepertoire page is redirected to VCAb, with the repertoire file from BRepertoire loaded.
Select the format of the repertoire file
VCAb support the following format of repertoire file: AIRR, Cell Ranger, and customized format. Example files for these three formats are provided, and user can view these files by clicking the corresponding link in the Upload repertoire table section on the web page.
For the customized file, it can be any repertoire file format, as long as it contains columns providing these information: amino acid sequence, chain type of the sequence (indicated by the value of IGH/IGK/IGL). Under the paired searching mode, column indicating the cell barcode should also be provided.
After the user upload the repertoire file & select the Customized option as the format, a panel below would be shown:
When the user select the Unpaired chains as the searching mode

When the user select the Paired H-L chains as the searching mode

Choose column(s) holding amino acid sequences:
- You can choose one or multiple column(s) holding amino acid sequence here. If multiple columns are chosen, the program will automatically concatenate sequence fragments in these columns into one sequence, and use this sequence for BLAST. This is useful if in the repertoire file, sequences are truncated into fragments of frameworks and CDRs, or fragments of V and C regions.
- Note: When multiple columns are selected, the concatenation of fragments depends on the order of the column names you input. For example, in the figure above, the sequence used for BLAST search would be fwr1 + cdr1 + fwr2 + cdr2 + fwr3 + cdr3 + fwr4.
Choose column indicating the identity of the chain (IGH/IGK/IGL):
- This column should indicate whether each chain is heavy or light chain. The value of this column can only be one of these: IGH, IGK, IGL. Otherwise the search can't be proceed, or the result is inaccurate.
Choose column indicating cell_id/barcode:
- This value is only needed in the paired search mode. This column should hold values unique for each cell (e.g. cell ID or barcode). If one cell contain only one H and one L chain, then the H-L chain is considered paired and will be inputted into the pipeline for searching.
In silico mutational scanning in VCAb provides the scanning for both V and C regions, with three methods: Rosetta point mutant scan for both V and C region, AntiBERTy for V region, and AlphaMissense for C region. For each wild-type (WT) residue on the sequence, its mutation to every one of the other 19 amino acids has been evaluated using these methods to yield a score measuring mutational impact.
All the results are shown in the format indicated in the figure below:
This results have two parts: the sequence banner indicating the residues on the pdb structure (Residue info is shown when the user hover onto it), and the heatmap containing the values indicating the mutational effect predicted by these three methods (Predicted mutational impact shown when the user hover onto it). The user can select the residues on the sequence banner to localize its position on 3D structural viewer.
Antibody Information panel contains four tabs, all of them are interactive with the 3D structural viewer:
- Antibody information
- Fab H-L contact map
- H-L interface residues
- Disulfide Bond
- In silico mutational scanning profiles
Antibody information

The Antibody Information tab displays the search results given the input specified in the Query panel. Central to the panel is a tabulated list of VCAb entries in the search output.
- Selecting the button 'Show' will display the selected VCAb structure in the structure viewer on the top right panel, and enable the other three tabs in the panel (Fab H-L contact map, H-L interface residues, Disulfide Bond) and tabs in the [[Interactive 3D Structure Viewer and Sequence Viewer]] panel.
- By default only some annotations are displayed in the table. You can search and select additional columns to be displayed (see below for a full description of the columns included in the database),
- If you query VCAb using the 'Sequence' tab in the Query panel, you will have additional functionalities to further filter the search results by isotype, structural coverage etc.
- You can download the search results as a zip file containing a comma separated value (CSV) file with the search results. Alternatively, you can select the option next to the button to download all entries in the search results in PDB format.
Description of columns
| Column name | Description |
|---|---|
| pdb | PDB identifier |
| Hchain | Chain identifier in the PDB entry corresponding to the Heavy chain. All chains encoding the same antibody are listed here. |
| Lchain | Chain identifier in the PDB entry corresponding to the Light chain. All chains encoding the same antibody are listed here. |
| heavy_vgene | The V gene of the heavy chain identified by BLAST against the V gene alleles for all the species |
| light_vgene | The V gene of the light chain identified by BLAST against the V gene alleles for all the species |
| antigen_chain | Chain identifier in the PDB entry corresponding to the antigen bound by the listed heavy-light chain pair. If multi-chain antigen, chain IDs are delimited by the pipe symbol. For antibody-only structures, this column will be empty. |
| antigen_type | Molecule type (protein / carbohydrate / nucleic acid ) of the corresponding antigen listed in antigen_chain. |
| antigen_description | Molecule name of the chain corresponding to the antigen. Empty if the structure does not contain antigen. |
| release-date | The 'DATE' entry in the PDB file. |
| resolution | Resolution in Angstrom of the structure. |
| method | Method of experimental structure determination |
| H_seq | Author-submitted sequence of the Heavy chain queried from the PDBe Application Programming Interface (API). |
| H_coordinate_seq | Sequence with ATOM coordinates found in the heavy chain in the PDB file. |
| L_seq | Author-submitted sequence of the Light chain queried from the PDBe Application Programming Interface (API). |
| L_coordinate_seq | Sequence with ATOM coordinates found in the light chain in the PDB file. |
| iden_code | Format: pdb_HL, where 'pdb' is the PDB identifier, and 'H' and 'L' are the first listed heavy and light chains in columns Hchain and Lchain for this given entry respectively. |
| Htype | Heavy chain isotype and allele for the best-scoring BLAST alignment between the CH sequence and the IMGT reference allele set. The percentage identity from BLAST is also given. |
| Ltype | Light chain type and allele for the best-scoring BLAST alignment between the CL sequence and the IMGT reference allele set. The percentage identity from BLAST is also given. |
| LSubtype | CL gene parsed from the column 'Ltype'. |
| Alternative_Htype | list of alignments to isotypes/alleles other than the one given in 'Htype', but with identical BLAST scores. |
| Alternative_Ltype | list of alignments to isotypes/alleles other than the one given in 'Ltype', but with identical BLAST scores. |
| carbohydrate | Full name of carbohydrate ligands found in the PDB structure. The associated chain identifiers are listed. |
| align_info | Details of the alignment for the allele given in 'Htype'. Format: [(region in H coordinate sequence), (region in reference H allele sequence), [CH domain, % of domain covered in structure, % of CH region in structure belonging to the domain) ]] |
| Domains in HC | A verbal description of the % covered statistics given in column 'align_info'. |
| Structural Coverage | Classification into 'Fab' or 'Full antibody'. |
| H_seq_VC_boundary | Position in author-submitted heavy chain sequence which marks the end of the VH domain and the beginning of the CH domain. |
| L_seq_VC_boundary | Position in author-submitted light chain sequence which marks the end of the VL domain and the beginning of the CL domain. |
| H_coordinate_seq_VC_boundary | Position in the heavy chain sequence parsed from PDB ATOM records, which marks the end of the VH domain and the beginning of the CH domain. |
| L_coordinate_seq_VC_boundary | Position in the light chain sequence parsed from PDB ATOM records, which marks the end of the VL domain and the beginning of the CL domain. |
| pdb_H_VC_Boundary | Residue number in the ATOM records of PDB corresponding to the heavy chain sequence which marks the end of the VH and the beginning of CH. Note this can be different from 'H_coordinate_seq_VC_boundary' if there are missing loops in the structure, or if the residue numbering in the PDB ATOM records contain insertion codes. |
| pdb_L_VC_Boundary | Residue number in the ATOM records of PDB corresponding to the light chain sequence which marks the end of the VL and the beginning of CL. Note this can be different from ':_coordinate_seq_VC_boundary' if there are missing loops in the structure, or if the residue numbering in the PDB ATOM records contain insertion codes. |
| HC_coordinate_seq | Sequence for the CH domain found in the ATOM records. |
| LC_coordinate_seq | Sequence for the CL domain found in the ATOM records. |
| HV_seq | Sequence for the VH domain parsed from the author-submitted sequence. |
| HC_seq | Sequence for the CH domain parsed from the author-submitted sequence. |
| LV_seq | Sequence for the VL domain parsed from the author-submitted sequence. |
| LC_seq | Sequence for the CL domain parsed from the author-submitted sequence. |
| disulfide_bond | Comma-separated list of pairs of cysteine residues involved in a disulphide bond. Format: [Residue 1](Chain ID 1)-[Residue 2](Chain ID 2): [C-alpha distance]. |
| elbow_angle | Calculation of the elbow angle (i.e. torsion angle between two pseudoaxes defined on the H and L chains in a Fab) performed following method described in Fernández-Quintero et al. Front Immunol 2020. |
| CH1-CL_interface_angle | Calculation of CH1-CL interface packing angle performed following method described in Fernández-Quintero et al. Front Immunol 2020. |
All the values of columns in italics are acquired from PDBeAPI
Fab H-L contact map
To enable this tab, one entry in the "Antibody Information" tab must be selected.
In this tab, a Fab contact map would be shown, with residues of heavy chain (numbered under IMGT scheme) as the x-axis, and residues of light chain as the y-axis. The map is colored by the distance between Cα, with the shortest distance as the darkest orange. Thus, two dark orange squares along the diagonal represent the contact map between VH-VL (lower left) and CH1-CL (upper right). The link to download the contact matrix is provided.
The button Hide non-interface residues would hide all the non-interface residues in the matrix and highlight only the H:L residue pairs if both residues are identified to be involved in H-L interface (Please see the demo below).
If you hover on the points on the contact map, the information about the H & L residues corresponding to this point would be shown, including the name of residue, IMGT numbering, distance between Cαs, and if both residues are involved in H-L interface. (Please see the demo below)

The Fab contact map is interactive with the 3D structural viewer. If the user drags a box on the contact map to select the residues, the structure shown in the 3D structure viewer would be automatically zoomed into the selected residues, with the interface residues highlighted and shown in ball-and-stick.

H-L interface residues
To enable this tab, one entry in the "Antibody Information" tab must be selected.
You can select the H-L interface residues in the table, and the corresponding residues would be interactively highlighted in the 3D structure viewer. Click the Zoom in to selected residues to zoom in to selected residues.

Disulfide Bond
To enable this tab, one entry in the "Antibody Information" tab must be selected.
You can select cysteine residues identified to be involved in disulfide bonds in the table, and the corresponding residues would be interactively highlighted in the 3D structure viewer. Click the Zoom in to selected residues to zoom in to selected residues.

In silico mutational scanning profiles
The in silico mutational scanning profiles are queryable for both heavy and light chains. Three methods are applied for this purpose: Rosetta point mutant scan, AntiBERTy, and AlphaMissense.
In this panel, three tabs are available:
Structure Viewer
To enable this tab, one entry in the "Antibody Information" tab in the [[Antibody Information]] panel must be selected. The structural viewer has two modes.
1. By default, only the H-L chain pair of the antibody indicated in the iden_code would be shown. You can rotate, zoom in/out the displayed structure.
2. By ticking the "View all the chains with the same PDB ID", the visualization of other chains is enabled, such as antigen chain(s) (if any) or other ligand(s) (if any). You can also tick the box ("Zoom in to the heavy-light chain pair of the selected VCAb entry") to focus on the H-L chain pair under this mode.
The coloring scheme and the option to download the structure are listed below the viewer.

The structure viewer is interactive with all the tabs in [[Antibody Information]] panel.
Sequence Coverage
To enable this tab, one entry in the "Antibody Information" tab in the [[Antibody Information]] panel must be selected.

Here, "atomic coordinate (HC)" refers to the sequence of heavy chain constant region, directly extracted from PDB file. "author-submitted sequence (HC)" is the heavy chain constant region sequence downloaded from PDBe API. And in the first row, the matched reference allele labeled with the domain (region) name is specified. In some cases, the sequence coverage for the atomic sequence and author-submitted sequence differs, like the example shown above (7c2l_HL).
In 7c2l_HL, the HC seq with atomic coordinate covers only CH1, while the author-submitted sequence (HC) covers all the domains of IgG1. One would classify the 7c2l_HL as full antibody if judged by author-submitted sequence (i.e. the sequence directly downloaded from PDBe API). Thus, in VCAb, coordinate sequence is applied for the structural coverage assignment, and 7c2l_HL would be correctly assigned as "Fab".
Sequence numbering
To enable this tab, one entry in the "Antibody Information" tab in the [[Antibody Information]] panel must be selected.

All the sequence is number under IMGT scheme (for both V and C regions). If you hover on the residue, the numbering information would be displayed. You can switch between heavy and light chain sequences by selecting options under Choose the sequence to display.
The process of the assembling of antibody structural space in VCAb is shown above. Please notice all the structures are experimental structures containing both V and C regions. During this process, several data sources/tools are applied:
PDB (Protein Data Bank)
PDB (Berman et al., 2003) is the data source for sequences and structures of antibodies. All the protein sequences are downloaded from worldwide PDB arcive, then antibody sequences are identified from them using ANARCI_vc. Structures of antibodies are downloaded using the modified shell script originally provided by RCSB PDB.
ANARCI_vc
ANARCI_vc is a package modified from ANARCI to enable both V and C numbering.
IMGT
IMGT (link here, Kaas et al.,2004, Ehrenmann and Lefranc,2011) serves as the data source for reference sequences due to its detailed information of alleles for different chain types, in the BLAST process for the identification of isotype, light chain type, and structural coverage.
The alleles downloaded from IMGT are collapsed based on if they have the same amino acid sequence. These unique allele amino acid sequences are collected into a fasta file(Please check them for details: unique H alleles, unique L alleles). These unique sequences form a customized BLAST database for VCAb sequences to blast against.
BLAST
BLAST Command Line is used to BLAST VCAb sequences against databases containing reference sequences, in order to identify the isotypes, light chain types and structural coverage of antibodies.
POPScomp
POPScomp is used to identify the CH1-CL interface residues. (POPScomp github, Publication: Kleinjung and Fraternali, 2005)
Mutational scanning tools
Please check In silico mutational scanning profile
Version: 19/08/2024
Documentation can be found in the following link:
VCAb github wiki
Users can search the VCAb by uploading repertoire files, entering the PDB identifiers, attributes (e.g. isotype, structural coverage, experimental methods, etc.), single sequence or sequences in batches. For each antibody entry, users can search for its sequence, isotype, structure and details of the CH1-CL interface. The structure and the CH1-CL interface residues of the antibody can be visualized and inspected in the web server. Researchers interested in antibody annotations and structures would benefit from the VCAb web server, especially due to the curated information it provides on isotype, light chain type and the CH1-CL interface residues.